Aidez-nous
Qu’est-ce que l’aplasie médullaire ?
L’aplasie médullaire est une maladie rare, dont l’origine est un dysfonctionnement de la moelle osseuse qui rend cette dernière incapable de produire des cellules sanguines (globules rouges, blancs et plaquettes) et donc de remplacer progressivement les cellules circulantes destinées à mourir naturellement.
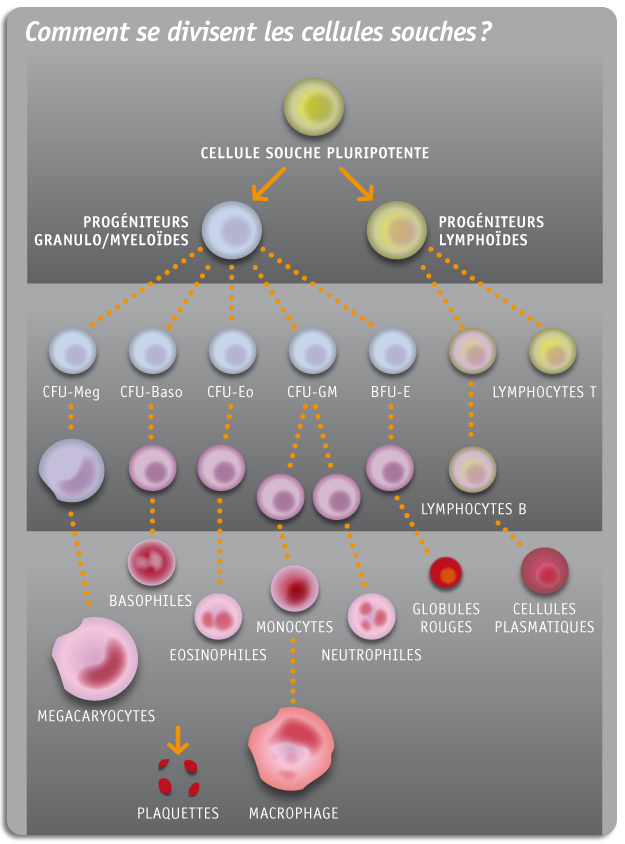
Dans la moelle osseuse, des cellules dites « souches » présentes en petite quantité, sont responsables de la production des cellules sanguines (globules rouges, blancs et plaquettes). Elles assurent le renouvellement quotidien de ces cellules (environ 100 milliards par jour chez un adulte).
Or dans l’aplasie médullaire ces cellules souches disparaissent, ne pouvant donc plus assurer leur rôle de renouvellement des cellules sanguines, dont la quantité va donc progressivement diminuer et de façon considérable.
L’aplasie est responsable du déficit d’un ou plusieurs types de cellules sanguines. Les conséquences de l’aplasie sont différentes en fonction du type de cellules déficitaires.
- Les globules rouges sont en charge du transport et de la distribution de l’oxygène dans l’organisme. Lors d’un déficit en globules rouges, l’anémie apparaît.
- Les globules blancs constituent le système de défense de notre corps contre les organismes étrangers (virus, bactérie, champignon,..). Lorsque leur nombre n’est pas suffisant, notre corps se défend moins bien et des infections peuvent apparaître
- Les plaquettes permettent la coagulation du sang et jouent un rôle primordial pour arrêter une hémorragie. Le déficit entraîne des troubles de la coagulation et par conséquent des hémorragies.
En fonction du type de lignées cellulaires atteintes les conséquences peuvent être multiples.
Quels symptômes et origines ?
Quelles sont les personnes touchées ?
Maladie rare avec une centaine de nouveaux cas par an, l’aplasie médullaire touche autant les hommes que les femmes. L’âge moyen est de 30 ans, cependant elle semble plus présente aux âges extrêmes de la vie, les enfants ou jeunes adultes et les personnes âgées.
Chez les jeunes adultes, il semblerait que les hommes soient plus atteints, alors que la population malade de plus 50 ans présente plus de femmes. L’aplasie est plus fréquente en Asie, qu’en Europe ou Amérique.
Quelle est son origine ?
Il s’agit d’une maladie acquise durant l’existence, elle n’est pas contagieuse. L’aplasie médullaire n’est donc pas une maladie héréditaire. Toutefois si certains gènes peuvent prédisposer à son développement, être porteur de ces gènes ne signifie pas forcément que l’on va développer la maladie. Ces gènes sont ainsi qualifiés de facteurs de susceptibilité, le gène HLADR2 a été identifié comme tel.
Il existe deux types d’aplasies médullaires : les formes acquises (>90% chez l’adulte) qui sont dans l’immense majorité des cas immunologiques (exceptionnelles formes toxiques) et les formes constitutionnelles ou génétiques au cours desquelles une anomalie génétique entraîne la disparition des cellules souches hématopoïétiques (jusqu’à 20% des formes pédiatriques). Les formes immunologiques se développent ainsi à n’importe quel moment de la vie, elles ne se transmettent pas à la descendance et ne sont pas contagieuses. Si des facteurs génétiques de prédisposition ont pu être identifié, qui favorisent leur survenue ils n’expliquent pas à eux seul la survenue de la maladie.
Le facteur déclenchant dans les formes acquises est inconnu. Si des formes de la maladie peuvent être distinguées – aplasie médullaire associée à un clone HPN, aplasies médullaires post hépatitiques, aplasies médullaires associées à des maladies auto-immunes ou aplasies médullaires idiopathiques quand rien ne les distingue – leurs mécanismes sont similaires.
Différents mécanismes contribuent vraisemblablement à la survenue d’une aplasie médullaire immunologique :
- Réaction immunitaire lymphocytaire anormale contre les cellules souches hématopoïétiques,
- Des anomalies intrinsèques aux cellules souches hématopoïétiques qui peuvent favoriser la survenue de cette réaction immunitaire,
- Un dysfonctionnement du microenvironnement médullaire nécessaire à la survie et à la maturation des cellules souches hématopoietiques,
- Des facteurs environnementaux susceptibles de stimuler cette réaction immunologique anormale
Quels sont les symptômes ?
L’aplasie médullaire peut apparaître brutalement ou alors progressivement et s’installer sans que le patient en ait conscience. En fonction du type de cellules sanguines touchées, les signes sont différents : fatigue liée à l’anémie, infections liées à la baisse des globules blancs ou saignements liés à la baisse des plaquettes.
Les globules rouges
L’anémie désigne le manque d’Hémoglobine dans le sang, qui entraîne un apport insuffisant en oxygène dans l’organisme. L’hémoglobine est contenue dans les globules rouges. Les signes de cette anémie sont :
- Pâleur de la peau, de la langue, des gencives et possible conjonctive (membrane qui recouvre le blanc de l’œil)
- Fatigue importante non réversible avec le repos
- Essoufflement, vertiges, palpitations cardiaques à l’effort
- Des douleurs musculaires possibles (sensations de muscles tétanisés)
- Peau terne et sèche, chute des cheveux et diminution de leur brillance et caractère soyeux
- Ongles cassants.
Les globules blancs
Fièvre inexpliquée ou infections à répétition sont le signe d’une diminution des polynucléaires neutrophiles. Ce sous type de globules blancs protège contre les infections bactériennes et à champignons, qui en quantité insuffisante ne peuvent plus assurer la défense de notre organisme. Les infections sont souvent des angines ou des furonculoses (infections de la peau caractérisées par la survenue de boutons purulents, ou furoncle).
Les plaquettes
Lorsque le nombre de plaquettes diminue, des saignements anormaux peuvent apparaître principalement au niveau du nez ou des gencives, ainsi que des bleus fréquents et inexpliqués (hématomes). Des hémorragies au niveau de la peau peuvent également apparaître, appelées purpura (petites taches d’un rouge violacé dont les dimensions varient d’une tête d’épingle à une lentille).
Quelle évolution?
Sans traitement, l’aplasie médullaire est imprévisible, mais les symptômes et la sévérité diffèrent d’un malade à l’autre.
Il arrive que les symptômes soient modérés et qu’une simple surveillance suffise. Le plus souvent, l’évolution se fait vers un état chronique où infections récidivantes et hémorragies de gravité variable se succèdent.
Ainsi, l’aplasie médullaire est responsable d’infections ou d’hémorragies pouvant mettre en péril le pronostic vital. Cependant, les récents progrès dans les traitements permettent d’améliorer nettement la longévité des patients.
D’autres maladies concomitantes ou à distance du diagnostic de l’aplasie peuvent également se déclarer.
C’est le cas de l’hémoglobinurie paroxystique nocturne (HPN) qui est présente chez 40 à 50 % des patients au diagnostic mais qui reste la plupart du temps asymptomatique. L’aplasie peut également évoluer vers des maladies tumorales de la moelle osseuse: myélodysplasie ou leucémie aiguë, mais celles-ci sont un peu très rares.
Dans quelques cas, une rémission spontanée peut survenir, au bout de quelques années d’évolution.
Quel diagnostic ?
Le diagnostic est prononcé lors de l’observation de signes cliniques particuliers :
- l’anémie : fatigue non compensée par le repos, pâleur de la peau, essoufflement, résistance à l’effort amoindrie, palpitations, vertiges, douleurs musculaires.
- des infections à répétition et/ou fièvre.
- des hémorragies : saignement du nez ou des gencives. (gingivorragies), saignements de la peau (purpura) ou muqueux, hématomes divers.
Le diagnostic de l’aplasie médullaire débute par l’exclusion d’autres causes possibles de la baisse des trois lignées sanguines (globules blancs, globules rouges et plaquettes), telles que les leucémies ou les syndromes myélodysplasiques. Cette étape repose notamment sur la réalisation d’un myélogramme, ou ponction médullaire.
Le diagnostic s’appuie sur plusieurs examens destinés à évaluer la quantité et la qualité des cellules sanguines, dans le sang périphérique comme dans la moelle osseuse :
-
La numération formule sanguine (NFS) : effectuée par une simple prise de sang, elle permet de mesurer le taux des différentes cellules sanguines (globules blancs, globules rouges et plaquettes).
-
La ponction médullaire : réalisée sous anesthésie locale, elle consiste à introduire une aiguille dans l’os iliaque (au niveau du bassin) ou le sternum afin de prélever un échantillon de moelle osseuse. Ce prélèvement permet d’évaluer la capacité de production cellulaire de la moelle et d’en analyser le fonctionnement.
-
La biopsie ostéo-médullaire : consiste à prélever, à l’aide d’une aiguille spécifique, un petit fragment osseux (ou « carotte ») au sommet du bassin. Il permet d’évaluer la richesse de la moelle osseuse et d’éliminer d’autres diagnostics, tels que la fibrose médullaire. Cette analyse contribue à confirmer le diagnostic d’aplasie médullaire.
La sévérité de l’aplasie médullaire s’évalue sur la numération formule sanguine réalisée avant toute transfusion.
La sévérité de la maladie est évaluée par les critères de Camitta :
- polynucléaires < 500 / mm3 (ou <0,5 10^9/l, chiffre sous lequel le risque infectieux devient grave
- plaquettes < 20 000 / mm3 ( ou <20 10^9/l), chiffre sous lequel le risque de saignement est important.
- Réticulocytes < 60 000 / mm3 (ou < 60 10^9/l)
Pour préparer la mise en place de traitements comme la transfusion sanguine ou la greffe de la moelle osseuse, des examens sanguins sont systématiquement effectués.
Le dépistage.
Il n’est pas possible de dépister l’aplasie médullaire avant l’apparition des premiers symptômes.
Quels traitements ?
Il existe deux types de traitements : les traitements de fond destinés à augmenter la production des cellules sanguines par la moelle osseuse et les soins de support pour éviter les conséquences de la baisse de production.
Les traitements de fonds
Il existe à ce jour deux types de traitements de référence pour l’aplasie :
La greffe de moelle osseuse et le traitement par immunosuppresseurs. Le choix du traitement est effectué en fonction des bénéfices et risques de chaque technique pour le patient.
Le traitement par immunosuppresseurs
Il s’agit de médicaments utilisés généralement dans le soin des maladies auto-immunes.
Le traitement par immunosuppresseurs peut être efficace (jusqu’à 80% de réponse), mais les résultats ne sont pas immédiats, un délai de 3 mois avant amélioration est généralement observé.
Le sérum antilymphocytaire (SAL) est le traitement de référence, associé à un autre immunosuppresseur la cyclosporine. Depuis plusieurs années, on associe chez les adultes et les adolescents, un stimulateur des cellules souches hématopoïétiques, l’eltrombopag, qui augmente la proportion et la vitesse de réponse.
En cas de contrindication au SAL (pathologies cardiovasculaires en particulier) l’eltrombopag peut être utilisé seul ou en association à la ciclosporine. Le taux de réponse de cette bithérapie est estimé à 50%.
Les patients qui n’ont pas répondu suffisamment pourront à nouveau être traités par immunosuppresseurs ou recevoir une allogreffe de cellules souches hématopoïétiques.
Complications et effets secondaires possibles.
Le risque principal du SAL est d’augmenter le risque d’infection pendant les premières semaines car l’association des immunosuppresseurs et les corticoïdes diminuent encore les défenses de l’organisme. Le déficit de globules blancs déjà présent doit être étroitement surveillé, le recours à la transfusion peut être nécessaire. Il peut également fragiliser la moelle osseuse et créer plus tard des complications. Les corticoïdes utilisés durant le premier mois pour éviter certaines réactions du SAL peuvent favoriser l’hypertension, le diabète, l’ostéoporose, les troubles du sommeil et la rétention hydro sodée.
D’autres effets secondaires peuvent également apparaître liés à l’utilisation des corticoïdes comme la :
- Cataracte
- Troubles du sommeil et/ou hormonaux
- Perte de masse musculaire
- ecchymoses
- troubles digestifs et prise de poids.
Des facteurs de croissance granulocytaire peuvent être associés au SAL et cyclosporine, ils stimulent la production de polynucléaires neutrophiles mais leur effet est inconstant.
La greffe de cellule souche hématopoïétique
La greffe est le seul traitement réellement curatif de l’aplasie médullaire. Il s’agit de remplacer la moelle osseuse du patient par celle d’un donneur compatible et de détruire les lymphocytes responsables de la maladie.
Elle est proposée en première intention choix chez les patients jeunes (moins de 40-50 ans) et ayant un donneur apparenté compatible (frère ou sœur).
En l’absence de donneur apparenté, la greffe n’est indiquée qu’en deuxième choix, une recherche est alors effectuée dans le fichier national ou international de donneurs. Au vu du risque de rechute et de dépendance à la ciclosporine, et au vu des bons résultats chez les plus jeunes, elle peut être cependant proposée au cas par cas en 1ere intention chez les moins de 18 ans.
La greffe nécessite de réaliser une chimiothérapie et expose à des complications immunologiques (réaction du greffon contre l’hôte (GVH) et rejet) dans 15% des cas mais la guérison à terme est obtenue dans 80 à 90% des cas.
Des greffes moins compatibles avec des donneurs à moitié identiques de la famille (haplo-identiques), des cellules souches issues du sang placentaires ou des donneurs du fichier non totalement identiques peuvent être proposées en l’absence de donneur compatible chez les jeunes.
Après la greffe…
Lorsqu’une greffe a été réalisée, le patient n’a pas encore récupéré un système immunitaire efficace et doit séjourner pendant plusieurs semaines en chambre stérile afin de limiter le risque d’infection.
Il est placé sous surveillance et notamment pour des risques hémorragiques, infectieux et anémiques.
La principale complication est la possibilité de rejet de greffe (10 % des cas), le système immunitaire du patient n’accepte pas les cellules de la greffe et les détruit. Celui-ci peut survenir rapidement ou dans les 2 à 3 années suivantes. Pour essayer de pallier ce problème, un traitement par immunosuppresseurs est prescrit (globulines anti-thymocytes, cyclosporine et cyclophosphamide).
Dans certains cas, les cellules greffées peuvent se retourner contre l’organisme du patient car il s’agit de cellules immunitaires qui ne reconnaissent pas les cellules du patient et veulent les détruire. On parle de réaction du greffon contre l’hôte, qui ne survient qu’en cas d’incompatibilité entre le donneur et le malade ou quand le patient a un système immunitaire très affaibli.
Traitements de support
Ces traitements permettent de prévenir ou de traiter les risques de l’aplasie :
- Risques liés à l’anémie : transfusion de culots de globules rouges pour maintenir le taux d’hémoglobine au-dessus de 8 g/dl
- Risque hémorragique : transfusion de plaquettes pour maintenir un taux supérieur à 10-20 000/mm3 (ou 10 à 20 G/l).
- Risques d’infection : tout épisode de fièvre, quand les polynucléaires neutrophiles sont < à 500 (ou < 0,5 G/l) nécessite une antibiothérapie adaptée.
Les facteurs de croissance permettent dans certains cas de stimuler le renouvellement et le développement des cellules sanguines.
Pour les globules rouges, différents types d’érythropoïétines peuvent être prescrits. Pour les globules blancs, il s’agit de protéines actives sur les granulocytes : G-CSF.
Le suivi
Le suivi de l’aplasie médullaire est effectué en milieu hospitalier en consultation d’hématologie et d’immuno-hématologie spécialisée.
Les unités de greffe de moelle assurent le suivi des personnes greffées.

La recherche
Actuellement de nombreuses équipes, en France et dans le monde, travaillent à une meilleure compréhension des processus de l’HPN et de l’aplasie médullaire ainsi qu’à l’amélioration de leurs traitements et de leur suivi à long terme.
Au niveau de la recherche fondamentale, de nouvelles cibles pour les traitements sont en cours d’investigation.
En parallèle de nombreuses études cliniques continuent d’être menées pour l’amélioration des traitements existants, afin de toujours mieux garantir leur sécurité et leur efficacité. Ainsi la greffe de sang placentaire constitue-t-elle désormais une sûre alternative aux greffes de moelle classiques. Un protocole de recherche clinique est actuellement en cours.
Observatoire national des insuffisances médullaires (RIME)
Les personnes atteintes d’aplasie médullaire, qu’elle soit acquise ou constitutionnelle, jouent un rôle central dans l’avancée des connaissances médicales. Chaque donnée clinique, chaque échantillon biologique prélevé est une pièce précieuse pour mieux comprendre la maladie, améliorer les prises en charge et développer de nouveaux traitements.
C’est dans cet esprit qu’a été créé en 2017 l’Observatoire national des insuffisances médullaires (RIME). Coordonné par le centre de référence (Pr Régis Peffault de Latour, Dr Flore Sicre de Fontbrune, Pr Thierry Leblanc, Mme Isabelle Brindel), cet observatoire permet la collecte anonymisée de données clinico-biologiques de patients atteints d’aplasie médullaire (acquise ou constitutionnelle) et d’hémoglobinurie paroxystique nocturne (HPN).
Pour enrichir cette base de données, une biobanque a également été mise en place sous la responsabilité du Pr Jean Soulier (Laboratoire d’hématologie, Hôpital Saint-Louis). Elle conserve des échantillons biologiques (sang, moelle osseuse, fibroblastes pour les formes constitutionnelles) prélevés à différentes étapes du parcours de soins.
En participant à ces projets de recherche, les patients contribuent activement à faire progresser la connaissance et à construire l’avenir des traitements. Pour en savoir plus sur les études en cours sur l’aplasie médullaire, cliquez ici.
HPN et l’aplasie
L’aplasie médullaire et l’HPN sont intrinsèquement liées. En effet, l’aplasie peut être une complication de l’HPN dans 20 à 30% des cas et l’HPN apparaît chez 30 % des patients traités par immunosuppresseurs.
L’hémoglobinurie paroxystique nocturne (HPN) est également une maladie rare, encore sous-diagnostiquée aujourd’hui. Il s’agit d’une maladie acquise durant la vie. Cette maladie n’est donc pas transmissible à la descendance. Elle n’est pas contagieuse non plus.
Elle touche principalement les adultes, aussi bien homme que femme mais peut survenir à tout âge. Elle est exceptionnelle avant 15 ans.
L’HPN se caractérise par une destruction chronique des globules rouges appelée hémolyse. Les principaux symptômes sont l’anémie, la fatigue, des urines foncées le matin et parfois des douleurs à l’abdomen ou au thorax, des difficultés à avaler, une dysfonction érectile… Le diagnostic se fait par une prise de sang
La fatigue est un des symptômes les plus invalidants car elle est souvent très intense au point de limiter la réalisation des activités quotidiennes des patients, de ralentir leur vie sociale et de provoquer des épisodes de somnolence dans la journée.
Mais le risque majeur pour des personnes atteintes d’HPN est la survenue de thromboses (caillots de sang) dont la formation silencieuse, entraîne un risque vital.
Les inhibiteurs du complément disponibles depuis 2007 permettent de bloquer la destruction des globules rouges, de prévenir la formation des caillots. Les patients ont désormais une survie presque identique à la population générale.
Documentations patient

Découvrez le livret patient « MON APLASIE ET MOI », un compagnon au quotidien pour mieux vivre avec l’Aplasie Médullaire
Vivre avec une aplasie médullaire acquise (anciennement dite idiopathique) représente un défi au quotidien. Pour vous accompagner, l’association HPN France – Aplasie Médullaire, en collaboration avec une hématologue et une membre bénévole de l’association, a contribué...

Le lien caché : Aplasie Médullaire et HPN
Dans le cadre de sa série de vidéos illustrées sous forme d’enquête, notre association publie aujourd’hui un nouvel épisode consacré au lien entre l’aplasie médullaire et l’HPN (Hémoglobinurie Paroxystique Nocturne). Ces deux maladies rares du sang sont étroitement...
Aplasie médullaire : comprendre la maladie autrement
Dans le cadre de sa nouvelle série de vidéos illustrées sous forme d’enquête, notre association publie aujourd’hui un premier épisode consacré à l’aplasie médullaire. Cette série pédagogique a pour objectif de mieux faire comprendre l’aplasie médullaire, l’HPN...

Parcours de soins et vécu : ce que disent vraiment les patients
La maladie rare ne se résume pas à un diagnostic, à des résultats biologiques ou à un protocole de traitement. Elle s’inscrit dans le quotidien, dans le corps, dans la tête, dans la vie sociale et professionnelle.C’est cette réalité-là que les patients atteints...

De l’« aplasie médullaire idiopathique » à l’« aplasie médullaire acquise » : comprendre l’évolution de la terminologie scientifique
Pendant longtemps, le terme « aplasie médullaire idiopathique » était utilisé pour désigner cette maladie rare.Aujourd’hui, les experts privilégient l’expression « aplasie médullaire acquise ». Ce changement n’est pas anodin : il reflète l’évolution des connaissances...

Fiche Orphanet Urgence : Bonnes pratiques en cas d’urgence – Aplasie Médullaire.
Un outil indispensable pour votre sécurité en situation d’urgence Les personnes atteintes d’Aplasie Médullaire font face à une maladie rare, complexe et parfois difficile à reconnaître en situation d’urgence, notamment lorsqu’elles sont prises en charge par des...
Publications médicales
Livret d’information pour les patients atteints d’Aplasie Médullaire
Notre Association HPN-AM a contribué à la rédaction du livret «Prise en charge d’une Aplasie Médullaire», publié en Octobre 2022, par le Centre de Référence des Aplasies Médullaires acquises et constitutionnelles. Vous y trouverez de nombreuses informations sur ces pathologie.
Pour le consulter, cliquez ici.
AUTRES PUBLICATIONS
Mise à jour du PNDS (Protocole National de Diagnostic et de Soins) sur les Aplasies Médullaires Acquises et Constitutionnelles (et l’HPN) – Mai 2023
Notre Association a participé à la mise à jour du PNDS, publié en Mai 2023, par le Centre de Référence des Aplasies Médullaires.
Pour lire ce long et précieux document, cliquer ici.
Aplasie médullaire sévère : nouvelle perspective dans la prise en charge thérapeutique – 6 janvier 2022
Le centre de référence des aplasies médullaires acquises et constitutionnelles coordonné par le Pr Régis Peffault de Latour, a évalué, dans le cadre d’une étude collaborative internationale, l’ajout d’un agoniste du récepteur de la thrombopoïétine (l’eltrombopag) au traitement standard immunosuppresseur.
Le traitement standard de l’aplasie médullaire sévère pour les patients non éligibles à une allogreffe de moelle osseuse associe depuis plus de 30 ans la ciclosporine et le sérum anti-lymphocytaire (traitement immunosuppresseur ciblant le système immunitaire) sans qu’aucun progrès n’ait pu être fait. Le taux de réponse attendu est d’environ 50%, ce qui signifie que la moitié des malades sont réfractaires au traitement.
Le travail récemment publié est une étude internationale multicentrique qui a évalué un agoniste du récepteur de la thrombopoïétine (l’eltrombopag) ajouté au traitement standard immunosuppresseur
« Cette association thérapeutique permet d’augmenter le taux de réponse d’environ 30% à six mois et devient ainsi le nouveau traitement de référence des patients adultes avec aplasie médullaire sévère au niveau international »
Pr Régis Peffault de Latour
Pour en savoir plus, c’est ici.
Résumé de la journée du CRMR Aplasie – Octobre 2020
=> Nouveaux traitements des Aplasies médullaires acquises (Pr de Latour)
1) Place de l’allogreffe avec un donneur non apparenté 10/10eme en 1ere ligne (en l’absence de donneur compatible familiale) chez les enfants et adolescent
2) Résultats de l’étude européenne RACE comparant l’association SAL de cheval, ciclosporine et eltrombopag au traitement de référence (SAL de cheval, ciclosporine)
3) Place des greffes haploidentiques (donneur intrafamilial à moitié compatible) chez les patients jeunes ayant des aplasies réfractaires au traitement immunosuppresseur
Pour lire les informations détaillées sur tous ces points cliquez ici.
=> Aplasie et Grossesse (Dr de Fontbrune)
Le lien entre grossesse et aplasie médullaire idiopathique pose question depuis de nombreuses années (le premier cas d’aplasie médullaire a été décrit chez une femme enceinte) : le lien exact n’est pas encore établi à ce jour. Plusieurs hypothèses existent :
1) la grossesse est un facteur déclenchant de l’aplasie,
2) la grossesse est un facteur aggravant de l’aplasie,
3) la grossesse est un moment propice aux hémogrammes qui favorise le diagnostic d’aplasie médullaire.
Pour lire la suite cliquez ici.
Cartes d’urgence pour l’Aplasie Médullaire – Juin 2018
Lors du congrès des urgences de juin 2018, la filière de santé des maladies rares immuno-hématologiques MaRIH a pu présenter, en compagnie des autres filières, les nouvelles cartes d’urgence maladies rares.
Ce document est destiné aux soignants aussi bien qu’aux patients, il informe les conduites à tenir en cas d’urgence. Il doit être complété par les médecins en charge du patient.
Les cartes sont désormais disponibles dans les centres de Reference MaRIH.
Voir la carte d’urgence pour les Aplasies Médullaires en cliquant ici
Contrat de collaboration entre l’AP-HP et Alexion
L’Assistance Publique-Hôpitaux de Paris a signé un contrat de collaboration de recherche translationnelle, pour quatre ans, avec la société de biotechnologie Alexion.
L’objectif : mieux comprendre les mécanismes impliqués dans la maladie du greffon contre l’hôte et l’aplasie médullaire, permettant de développer de nouveaux traitements contre ces deux graves maladies rares.
La réaction du greffon du donneur contre l’hôte est l’une des complications majeures survenant après une greffe de cellules souches hématopoïétiques (CSH), un type de cellules sanguines. Sa fréquence est de l’ordre de 50 à 60% des greffes de CSH et elle est sévère, voire mortelle, dans plus de 10% des cas.
L’aplasie médullaire est une maladie rare caractérisée par la destruction de la moelle osseuse des patients par leur propre système immunitaire. 150 nouveaux cas sont recensés en France chaque année.
Afin d’améliorer la compréhension et le traitement de ces deux pathologies, l’AP-HP a donc conclu pour le compte de l’hôpital Saint-Louis, AP-HP un partenariat, avec la société Alexion, qui développe et commercialise des traitements pour les maladies graves rares et ultra-rares.
Le projet « AP-Alexion » porté par un consortium de médecins et de chercheurs, bénéficiera de l’existence à l’hôpital Saint-Louis de compétences de renommée internationale en hématologie, hémato-biologie et bio-statiques.
Il aura pour objectif d’assurer un transfert rapide des connaissances entre les sphères de la recherche et du soin, et ainsi de faciliter l’émergence de nouveaux diagnostics et traitements.
Pour le Pr Gérard Socié*, l’ambition de ce projet qu’il coordonne avec le Pr Régis Peffault de la Tour* est « celle d’une recherche collaborative de niveau international, dont le but ultime est la compréhension des mécanismes intimes des pathologies et le développement de nouvelles armes thérapeutiques ».
Le Projet « AP-Alexion » s’inscrit parfaitement dans les initiatives prises par l’hôpital Saint-Louis pour développer et améliorer la prise en charge des patients atteints de maladies rares immuno-hématologiques. Trois centres de référence nationaux et une filière de santé maladies rares immuno-hématologiques (maRIH) y sont déjà regroupés.
Son service d’hématologie-greffe, qui est aussi le centre de référence maladie rare pour les aplasies médullaires, a fait du traitement de la maladie du greffon contre l’hôte un axe majeur de sa recherche. C’est le centre qui réalise actuellement le nombre de greffes CSH le plus important en France (130 sur les 2000 greffes réalisées à l’échelle nationale).
Avec ce nouveau partenariat, l’AP-HP renforce également ses liens avec la société Alexion, déjà impliquée dans un projet de l’Institut Imagine situé à l’hôpital Necker-Enfants malades, AP-HP.
* service d’hématologie-greffe et centre de référence maladie rare – aplasies médullaires de l’hôpital Saint-Louis, AP-HP
Voir le communiqué de l’AP-HP du 19/09/2016 http://www.aphp.fr/contenu/lap-hp-et-alexion-concluent-un-partenariat-de-recherche-sur-deux-maladies-rares-du-sang